现在,二十多年过去了,MIT教授Jonathan Weissman及其同事已经超越了序列并提出了人类细胞中表达的基因的第一个全面功能图。这个项目的数据于6月9日在线发表在《Cell》上,其将每个基因跟它在细胞中的工作联系起来,这代表了多年来在单细胞测序方法Perturb-seq上合作的结晶。
这些数据可供其他科学家使用。Weissman说道:”这是一个很大的资源,就像人类基因组是一个很大的资源一样,你可以进去做基于发现的研究。你不必提前定义你要研究的生物学,你有这个基因型-表型关系图,你可以进去筛选数据库,而不必做任何实验。”据悉,他还是怀特海研究所的成员和霍华德-休斯医学研究所的调查员。
这使得研究人员得以深入研究各种生物学问题。他们用它来探索功能未知的基因对细胞的影响、研究线粒体对压力的反应并筛选出导致染色体丢失或增加的基因,这种表型在过去被证明很难研究。“我认为这个数据集将使那些来自生物学其他领域的人能够进行各种我们甚至还没有想到的分析,突然间他们就有了这个可以利用的东西,”这项研究的论文共同第一作者、前魏斯曼实验室博士后Tom Norman说道。
开创性Perturb-seq方法
该项目利用的Perturb-seq方法使得科学家们有可能以前所未有的深度跟踪开启或关闭基因的影响。这种方法由包括魏斯曼和MIT教授Aviv Regev在内的一组研究人员于2016年首次发表,不过它只能用于小规模的基因集且花费巨大。
大量的Perturb-seq图谱是由Joseph Replogle的基础工作促成,他是Weissman实验室的医学博士生,也是本论文的第一作者之一。Replogle与Norman、Britt Adamson(普林斯顿大学分子生物学系的助理教授)及10x基因组学的一个小组合作着手创建一个可以扩大规模的新版Perturb-seq。研究人员于2020年在《Nature Biotechnology》上发表了一篇概念验证的论文。
Perturb-seq方法使用CRISPR-Cas9基因组编辑将遗传变化引入细胞,然后使用单细胞RNA测序来捕获有关因特定遗传变化而表达的RNA的信息。由于RNA控制着细胞行为方式的所有方面,这种方法可以帮助解读基因变化的许多细胞效应。
自他们最初的概念验证论文以来,Weissman、Regev及其他人已经在更小的范围内使用这种测序方法。如研究人员在2021年使用Perturb-seq来探索人类和病毒基因在感染HCMV(一种常见的疱疹病毒)的过程中如何互动。
在新研究中,Replogle和包括Weissman实验室的研究生和该论文的共同第一作者Reuben Saunders在内的合作者将该方法扩大到整个基因组。他使用人类血癌细胞系以及来自视网膜的非癌细胞、对超过250万个细胞进行了Perturb-seq并利用这些数据建立了一个将基因型和表型联系起来的全面地图。
深入研究数据
在完成筛选后,研究人员决定将他们的新数据集投入使用并着手研究一些生物学问题。Tom Norman指出:“Perturb-seq的优势在于它可以让你以一种无偏见的方式获得一个大数据集。没有人完全知道你能从这种数据集中得到的极限是什么。现在问题是,你实际上用它做什么?”
第一个最明显的应用是研究具有未知功能的基因。因为该屏幕也读出了许多已知基因的表型,研究人员可以用这些数据来比较未知基因和已知基因并寻找类似的转录结果,这可能表明这些基因产品作为一个更大的复合物的一部分一起工作。
一个名为C7orf26的基因的突变尤其引人注目。研究人员注意到,那些被移除导致类似表型的基因是一种叫做Integrator的蛋白质复合物的一部分,该复合物在创造小核糖核酸方面起着作用。Integrator复合体由许多较小的亚单元组成--以前的研究表明有14个单独的蛋白质--而研究人员能确认C7orf26构成了该复合体的第15个组成部分。
他们还发现,这15个亚单位在较小的模块中一起工作并在整合者复合物中执行特定的功能。Saunders说道:“如果没有这种千尺高楼平地起的情况,这些不同的模块在功能上是如此不同,这一点并不清楚。”
而Perturb-seq的另一个好处是,由于该检测专注于单细胞,研究人员可以使用这些数据来观察更复杂的表型,当它们跟来自其他细胞的数据一起研究时这些表型会变得模糊不清。“我们经常把‘基因X’被敲除的所有细胞拿出来,把它们平均在一起,然后看看它们如何变化,”Weissman介绍称,“但有时当你敲除一个基因时,失去同一基因的不同细胞会有不同的行为,而这种行为可能会被平均数所忽略。”

相关资料图
研究人员发现,一个基因子集,其去除导致不同细胞的不同结果,负责染色体的分离。它们的移除导致细胞失去一条染色体或捡到一条额外的染色体,这种情况被称为非整倍体。“你无法预测失去这个基因的转录反应是什么,因为它取决于你获得或失去什么染色体的次级效应,”Weissman介绍道,“我们意识到,我们可以扭转这种情况,创造这种复合表型,寻找染色体获得和失去的特征。通过这种方式,我们已经完成了第一个全基因组范围的筛选以寻找DNA正确分离所需的因素。”
“我认为非整倍体研究是迄今为止这一数据的最有趣的应用。它抓住了一个你只能用单细胞读出的表型。你不能用其他方式去追求它,”Norman说道。
研究人员还利用他们的数据集来研究线粒体如何对压力做出反应。从自由生活的细菌进化而来的线粒体,在其基因组中携带13个基因。在核DNA内,约有1000个基因跟线粒体功能有某种程度的关系。Replogle说道:“人们对核DNA和线粒体DNA在不同的细胞条件下是如何协调和调节的,特别是当一个细胞受到压力时,已经关注了很久了。”
研究人员发现,当他们扰乱不同的线粒体相关基因时,核基因组对许多不同的遗传变化作出类似的反应。然而线粒体基因组的反应要多得多。
Replogle指出:“线粒体为什么仍有自己的DNA,这仍然是一个开放的问题。从我们的工作中得到的一个大的启示是,拥有独立的线粒体基因组的一个好处可能是对不同的压力源有局部的或非常具体的遗传调节。”
“如果你有一个线粒体被破坏,而另一个线粒体以不同的方式被破坏,这些线粒体可能会做出不同的反应,”Weissman说道。
在未来,研究人员希望将Perturb-seq用于他们开始使用的癌细胞系以外的不同类型的细胞。另外,他们还希望继续探索他们的基因功能图并希望其他人也能这样做。Norman说道:”这确实是作者和其他合作者多年工作的结晶,我真的很高兴看到它继续成功和扩大。”
-

每日动态!发力食品科研,郑州大学携手好想你共建营养健康食品研究院
头条 22-06-12
-

当前聚焦:知网开放个人查重服务:千字1.5元,研究生学位论文免费查3次
头条 22-06-12
-

每日快讯!河南省昨日零新增
头条 22-06-12
-

每日热文:3天涨粉超130万!俞敏洪直播火出圈,不仅卖货还教英语!股价飙升
头条 22-06-12
-

快资讯丨券商“补血”进行时 年内发债超4600亿
头条 22-06-12
-

今热点:从屠宰到深加工,雨轩股份打开牛羊肉新“食”尚 | 预制菜看原阳②
头条 22-06-12
-

每日短讯:河南省水利厅党组成员申季维:2022年年底前基本完成贾鲁河主体工程等项目
头条 22-06-12
-

焦点!拓展欧洲朋友圈!中德柔性电子项目合作签约仪式在郑举行
头条 22-06-12
-

信息:春雪食品:已被京东纳入预制菜战略扶持品类
头条 22-06-12
-

信息:泰格生物、和泽医药、裕松源药业三方在漯河签约
头条 22-06-12
-

今日关注:财政部、应急管理部紧急预拨3.6亿元,支持地方做好防汛抗旱救灾工作
头条 22-06-12
-

视点!河南铁建投集团签订河南首个土地综合开发实施协议
头条 22-06-12
-

今日热门!滴滴退市 股份将转到OTC场外交易市场进行交易
头条 22-06-12
-
一个月暴涨3000亿,比亚迪开启"狂暴模式"!巴菲特大赚38倍,广州神秘富豪一战成名
头条 22-06-11
-
河南昨日无新增本土确诊病例
头条 22-06-11
-
11户央企14名领导人员职务任免 | 全名单
头条 22-06-11
-
隔夜欧美·6月11日
头条 22-06-11
-
立方风控鸟·早报(6月11日)
头条 22-06-11
-
立方风控鸟·晚报(6月10日)
头条 22-06-11
-
河南成功发行第八批政府债524.8079亿 全场认购倍数达24.21倍
头条 22-06-11
-
华晨集团重整草案出炉
头条 22-06-11
-
杭州银行被罚40万元
头条 22-06-11
-
江苏银行被罚
头条 22-06-11
-
济源现代物流集团挂牌成立
头条 22-06-10
-
新增两家,南阳国家级双创载体数量居全省第一方阵
头条 22-06-10
-
融通地产、河南航投等企业联合到许昌考察,就片区综合开发、物流园等进行对接
头条 22-06-10
-
国家统计局:5月份PPI同比上涨6.4% 环比上涨0.1%
头条 22-06-10
-
漯河大专学历及技能人才购房补贴1万元
头条 22-06-10
-
中央汇金出手!将入股华融湘江银行
头条 22-06-10
-
立方风控鸟·早报(6月10日)
头条 22-06-10
-
河南昨日本土零新增!
头条 22-06-10
-
抢占发展新高地 河南智慧终端联合实验室在南阳社旗县启动运行
头条 22-06-10
-
河南前5个月省重点项目完成投资8234亿元 占年度目标的60.9%
头条 22-06-10
-
何雄会见北京联东集团常务副总裁梁环宇 助力郑州塑造产业新优势
头条 22-06-10
-
隔夜欧美·6月10日
头条 22-06-10
-
郑州组织青年人才购房专题推介会 3家央企推出八折优惠现房房源
头条 22-06-10
-
总投资32亿元!鹤壁云签约4个大项目
头条 22-06-10
-
郑州绿地JW万豪酒店要卖?消息人士:有意向但最终实施须绿地总部批准
头条 22-06-10
-
立方风控鸟·晚报(6月9日)
头条 22-06-10
-
一季度保险行业总体净利润同比下降65.5%
头条 22-06-10
-
金刚退:聘请渤海证券为新三板主办券商
头条 22-06-10
-
强对流来袭 河南发布山洪灾害预警
头条 22-06-10
-
最新!全国一二线楼市新政图鉴
头条 22-06-09
-
规模50亿元!国家中小企业发展基金子基金落户成都
头条 22-06-09
-
央行今日进行100亿元7天期逆回购操作
头条 22-06-09
-
李跃勇会见新华水力发电董事长张焰一行,双方就推进能源开发合作等达成共识
头条 22-06-09
-
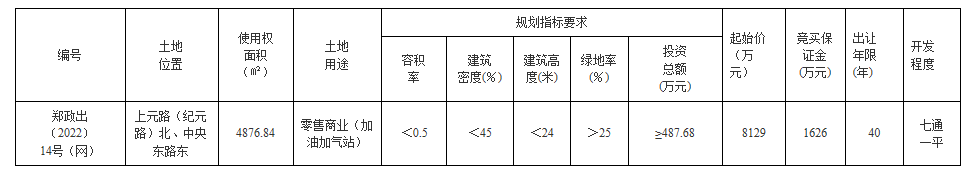
起始价7.4亿元!商丘拟出让两宗196亩居住用地
头条 22-06-09
-
河南昨日新增本土无症状感染者1例
头条 22-06-09
-
何雄会见中兴通讯执行副总裁一行,拓展智慧城市、数字经济等领域合作
头条 22-06-09
-
隔夜欧美·6月9日
头条 22-06-09
-
立方风控鸟·早报(6月9日)
头条 22-06-09
-
皮海洲:美女分析师喊出4000点 投资者小心被误导
头条 22-06-09
-
立方风控鸟·晚报(6月8日)
头条 22-06-09
-
一粒小麦 七十二变 | 极刻
头条 22-06-09
-
郑州举行“双碳”技术能力发布会,清华大学专家团队领衔
头条 22-06-09

















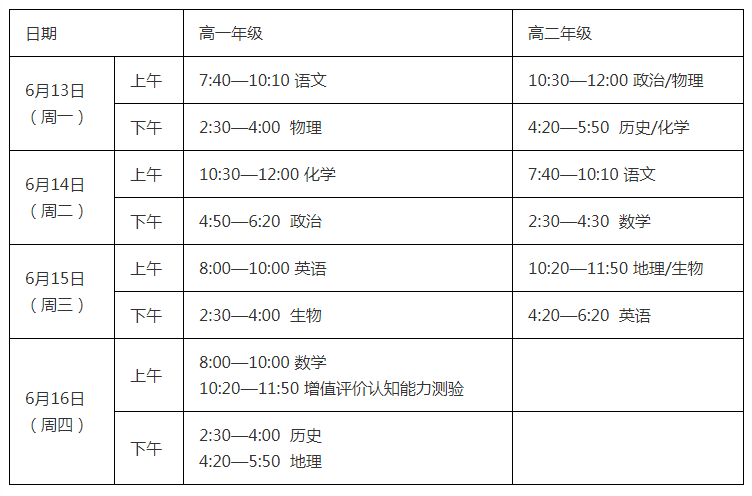








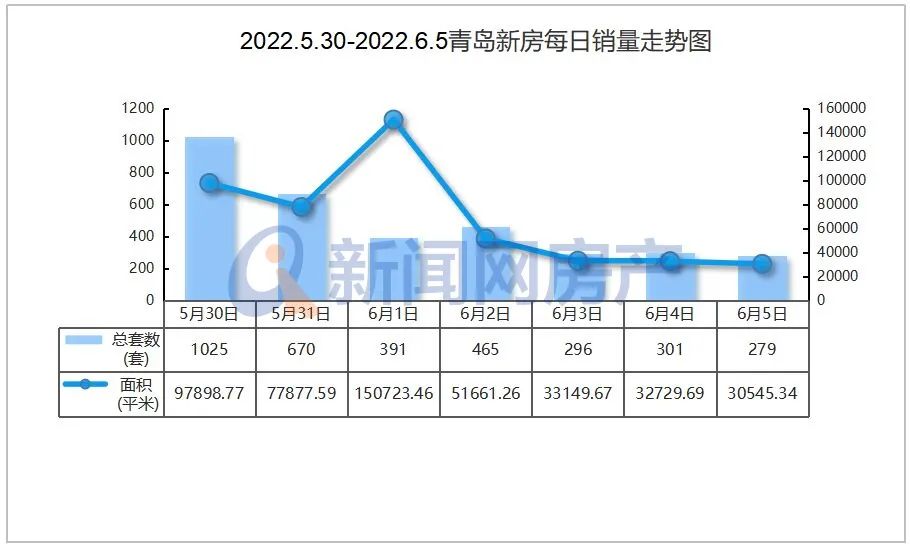





















































- 百事通!@各位高考生:军校在豫招生计划请2022-06-13
- 动态:新综合地图将每个人类基因跟它的功能2022-06-13
- 热资讯!星火成炬|闪光少年2022-06-13
- 滚动:人民日报:“中国节日”系列节目引领2022-06-13
- 每日速读!警方通报唐山蛋糕店被敲诈勒索事2022-06-13
- 微速讯:革命性大脑成像将有望揭开阿尔茨海2022-06-13
- 滚动:“超级月亮”14日现身夜空,系本年度2022-06-13
- 即时看!上外通报一学生被投放异物:涉事学2022-06-13
- 【独家】云南临沧市耿马县发生3.1级地震,2022-06-13
- 每日时讯!河南102岁曲艺“活化石”再登舞台2022-06-13
- 快报:唐山女子举报遭酒吧老板拘禁,警方:2022-06-13
- 每日关注!唐山打人案9名嫌犯已被廊坊警方逮2022-06-13
- 前沿热点:重点整治故意伤害、侮辱妇女等行2022-06-13
- 速看:俄罗斯航天局局长:宇宙中或存在比地2022-06-13
- 每日聚焦:美国各地民众集会要求政府应对枪2022-06-13
- 今日热讯:水利部针对南方雨情再启洪水防御2022-06-13
- 每日报道:三星堆发现近1米高青铜神坛 新2022-06-13
- 麻袋财富:信息技术为人们带来便捷生活,互2022-06-12
- 每日动态!发力食品科研,郑州大学携手好想2022-06-12
- 当前聚焦:知网开放个人查重服务:千字1.52022-06-12
- 观焦点:广西北流新丰镇洪涝消退 危险地段2022-06-12
- 热门:福建发布山洪灾害风险橙色预警2022-06-12
- 今日视点:国家卫健委:昨日新增本土确诊病2022-06-12
- 天天资讯:本土新增122+74,其中内蒙古78+82022-06-12
- 热议:福建昨日新增本土无症状感染者1例2022-06-12
- 新消息丨注意!河南将停考4个高等教育自考2022-06-12
- 当前焦点!NASA正在发射6颗新卫星以帮助研究2022-06-12
- 观焦点:卫星图像展示EF-3龙卷风给密歇根州2022-06-12
- 每日视讯:文化和自然遗产日:我国世界自然2022-06-12
- 今日热闻!第六个“中国器官捐献日”,黄梅2022-06-12














